ĐỊNH NGHĨA
Suy giảm miễn dịch nguyên phát là bệnh di truyền có thể liên quan đến mọi khía cạnh của đáp ứng miễn dịch, từ bẩm sinh cho đến đáp ứng, cũng như biệt hóa tế bào, bộ phận tác động chức năng, và điều chỉnh miễn dịch (Bảng 168-1). Hậu quả của suy giảm miễn dịch nguyên phát thay đổi rất rộng như sự khiếm khuyết chức năng của các phân tử và bao gồm tổn thương bị nhiễm trùng do nhiễm trùng cơ hội và gây bệnh, các phản ứng miễn dịch như dị ứng, tăng sinh tế bào lympho, và tự miễn dịch, và tăng nguy cơ ung thư. Khu vực và vị trí nhiễm trùng và các vi sinh vật gây bệnh thường giúp cho bác sĩ trong chẩn đoán.
CHẨN ĐOÁN (XEM BẢNG 168-2)
PHÂN LOẠI (BẢNG 168-1)
SUY GIẢM CỦA HỆ MIỄN DỊCH BẨM SINH
Chiếm ~10% của tất cả thiếu hụt miễn dịch nguyên phát (Bảng 168-1).
SUY GIẢM CỦA HỆ MIỄN DỊCH ĐÁP ỨNG
Hội chứng suy giảm tế bào lympho T
Nhóm suy giảm miễn dịch kết hợp nặng (SCID) của suy giảm miễn dịch nguyên phát hiếm đặc trưng bởi một khối sâu sắc trong sự phát triển của tế bào T như là hậu quả của sự thiếu hụt nội tại. Hậu quả lâm sàng xảy ra trong vòng 3–6 tháng sau sinh. Các biểu hiện lâm sàng thường gặp nhất là tái phát nấm candida miệng, chậm phát triển, tiêu chảy kéo dài, nhiễm Pneumocystis jiroveci. Sáu cơ chế gây bệnh riêng biệt đã được xác định:
BẢNG 168-1 PHÂN LOẠI CÁC BỆNH SUY GIẢM MIỄN DỊCH NGUYÊN PHÁT
Suy giảm của hệ miễn dịch bẩm sinh
• Tế bào thực bào
– Giảm sản xuất: giảm bạch cầu bẩm sinh nặng (SCN)
– Không có lách
– Giảm bám dính: giảm bám dịch bạch cầu (LAD)
– Giảm tiêu diệt: bệnh u hạt mạn tính (CGD)
• Các thụ thể miễn dịch bẩm sinh và sự truyền tín hiệu
– Giảm tín hiệu thụ thể Toll-like
– Nhạy cảm với bệnh do mycobacterium theo quy luật Menden
• Giảm bổ thể
– Con đường cổ điển, thay thế và lectin
– Chu trình tan
Suy giảm của hệ miễn dịch đáp ứng
| Tế bào lympho T – Giảm phát triển – Giảm tồn tại, di cư, chức năng |
Suy giảm miễn dịch kết hợp trầm trọng (SCIDs) hội chứng DiGeorge Suy giảm miễn dịch kết hợp trầm trọng Hội chứng Hyper-IgE (tính trạng trội trên NST thường) Thiếu phối tử CD40 Hội chứng Wiskott-Aldrich Thất điều-giãn mạch và thiếu sót sửa chữa ADN khác |
| Tế bào lympho B – Giảm phát triển – Giảm chức năng |
Chứng không có gamma globulin máu di truyền trên NST giới tính X và di truyền lặn NST thường Hội chứng siêu IgM Suy giảm miễn dịch thay đổi thường gặp (CVID) thiếu IgA |
Các suy giảm điều tiết
| • Miễn dịch bẩm sinh | Hội chứng tự viêm (ngoài phạm vi của chương này) Viêm đại tràng nặng |
| • Miễn dịch đáp ứng | Hội chứng thực bào tế bào máu (HLH) Hội chứng tăng sinh tế bào lympho tự miễn (ALPS) Các bệnh tự miễn và bệnh viêm (IPEX, APECED) |
Các từ viết tắt: APECED: một bệnh tự miễn di truyền đặc trưng bởi ít nhất 2 trong 3 đặc điểm: suy cận giáp, nhiễm nấm candida, suy thượng thận; IPEX: một bệnh hiếm gặp liên quan đến sự rối loạn chức năng của thụ thể sao mã FOXP3, được coi là thụ thể điều hòa chính của tế bào điều hòa dòng T.
BẢNG 168-2 TEST THƯỜNG ĐƯỢC DÙNG NHẤT ĐỂ CHẨN ĐOÁN SUY GIẢM MIỄN DỊCH TIÊN PHÁT (PID)
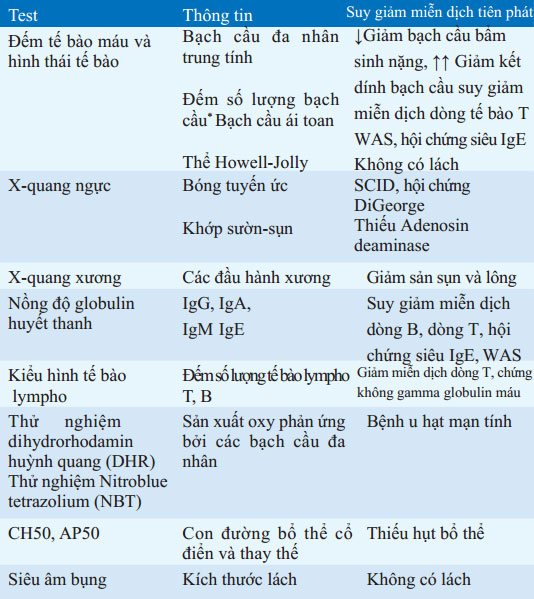
*Số lượng bình thường thay đổi theo tuổi. Ví dụ, số lượng tế bào lympho từ 3000 đến 9000/μl máu khi dưới 3 tháng và từ 1500 đến 2500/μl ở người lớn.
Các từ viết tắt: SCID: suy giảm miễn dịch kết hợp trầm trọng; WAS: hội chứng Wiskott-Aldrich.
- Thiếu tín hiệu cytokin: suy giảm miễn dịch kết hợp trầm trọng thường gặp nhất chiếm 40–50% các trường hợp với sự vắng mặt của tế bào T và tế bào diệt tự nhiên. Những bệnh nhân này có sự thiếu hụt trong chuỗi thụ thể gamma được chia sẻ bởi nhiểu thụ thể cytokin (interleukin 2, 4, 7, 9, 15, 21). Kiểu hình giống nhau được thấy ở suy giảm miễn dịch kết hợp
trầm trọng di truyền theo NST X được di truyền như một bệnh di truyền lặn trên NST thường do đột biến ở gen JAK3 protein kinase. - Thiếu hụt chuyển hóa purin: có khoảng 20% bệnh nhân suy giảm miễn dịch kết hợp trầm trọng bị thiếu adenosin deaminase (ADA) do đột biến ở gen ADA.
- Thiếu hụt trong sắp xếp của thụ thể tế bào T và B: chiếm khoảng ~20–30% trường hợp của suy giảm miễn dịch kết hợp trầm trọng. Những thiếu hụt chính liên quan đến kích hoạt gen tái tổ hợp ADN (RAG-1, RAG-2) phụ thuộc protein kinase, ADN 4, và thiếu hụt Cernunnos (tật đầu nhỏ, suy dinh dưỡng bào thai, chậm phát triển nghiêm trọng và mặt giống chim).
- Thiếu (tiền) thụ thể tế bào T truyền tín hiệu trong tuyến ức: thiếu sót hiếm ở tiểu đơn vị CD3 liên quan với (tiền) TCR và CD45.
- Loạn sinh lưới: cực kỳ hiếm. Kết quả từ thiếu adenylat kinase 2.
- Thiếu hụt ở đường ra của tế bào lympho: thiếu hụt ở đường ra của tế bào T từ tuyến ức là kết quả của sự thiếu hụt ở coronin-1A.
ĐIỀU TRỊ Suy giảm miễn dịch kết hợp trầm trọng
Điều trị dựa trên cấy ghép tế bào gốc tạo máu (HSCT).
Những suy giảm miễn dịch nguyên phát liên quan đến tế bào T khác
- Hội chứng DiGeorge: phát triển không bình thường của tuyến ức.
- Hội chứng siêu IgE.
- Thiếu phối tử CD40.
- Hội chứng Wiskott-Aldrich.
- Thất điều-giãn mạch và các thiếu hụt sửa chữa ADN khác
ĐIỀU TRỊ Những suy giảm miễn dịch nguyên phát tế bào T khác
Điều trị phức tạp và phần lớn đang được nghiên cứu. Cấy ghép tế bào gốc tạo máu giữ một vai trò ở một vài bệnh. Các vắc xin sống và truyền máu có chứa các tế bào T hoạt động nên tránh hoàn toàn. Dự phòng cho bệnh viêm phổi do Pneumocystis jiroveci nên được xem xét ở những bệnh nhân có thiếu hụt tế bào T trầm trọng.
Hội chứng suy giảm tế bào lympho B
Những suy giảm ảnh hưởng đến tế bào B là suy giảm miễn dịch nguyên phát hay gặp nhất và chiếm ~60–70% các trường hợp. Suy giảm sản xuất kháng thể đưa đến nhiễm khuẩn sinh mủ xâm lấn cũng như tái phát nhiễm trùng ở xoang và phổi. Toàn bộ Suy giảm sản xuất kháng thể (không có gamma globulin máu) dẫn đến nhiễm enterovirus lan tỏa gây viêm màng não, viêm gan và bệnh giống viêm da cơ. Chẩn đoán dựa vào việc xác định nồng độ 1g.
• Không có gamma globulin máu: do đột biến ở NST X của gen Bruton’s tyrosine kinase (Btk)trong 85% trường hợp.
• Siêu IgM: ở phần lớn bệnh nhân hội chứng này là kết quả do ảnh hưởng của di truyền liên kết NST X ở gen mã hóa phối tử CD40. Bệnh nhân có IgM bình thường hoặc tăng với nồng độ thấp hoặc không có IgG và IgA.
• Suy giảm miễn dịch thông thường (CVID): Nhóm hỗn tạp của hội chứng đặc trưng bởi nồng độ thấp của một hoặc nhiều lớp Ig trong huyết thanh.
Tỉ lệ ước tính là 1/20.000 người. Bên cạnh nhiễm trùng, bệnh nhân có thể có tăng sinh tế bào lympho, tổn thương u hạt, viêm đại tràng, bệnh tự miễn dịch qua trung gian kháng thể, và u lympho.
• Thiếu IgA chọn lọc: là suy giảm miễn dịch phổ biến nhất; ảnh hưởng đến 1/600 người. Đa số các cá nhân bị ảnh hưởng không tăng nhiễm trùng; các kháng thể kháng IgA có thể gây sốc phản vệ trong quá trình truyền máu hoặc huyết tương; có thể tiến triển đến CVID.
• Thiếu kháng thể chọn lọc với các kháng nguyên polysaccharid.
ĐIỀU TRỊ Hội chứng suy giảm tế bào B/globulin miễn dịch
Áp dụng globulin miễn dịch đường tĩnh mạch (chỉ dành cho những bệnh nhân có bệnh nhiễm khuẩn tái phát và thiếu hụt IgG):
• Bắt đầu với liều 400–500 mg/kg mỗi 3–4 tuần
• Chỉnh liều để giữ đỉnh nồng độ IgG mức 800 mg/dl
• Tiêm dưới da 1 lần/tuần có thể được xem xét ở một số bệnh nhân.
SUY GIẢM ĐIỀU TIẾT
Hiếm nhưng ngày càng định rõ tính chất suy giảm miễn dịch nguyên phát gây rối loạn điều hòa cân bằng nội môi của hệ thống miễn dịch hoặc một mình hoặc kết hợp với tăng tính dễ bị tổn thương dẫn đến nhiễm trùng (Bảng 168-1).
